Sickle Cell Anemia: What to Know and How to Manage
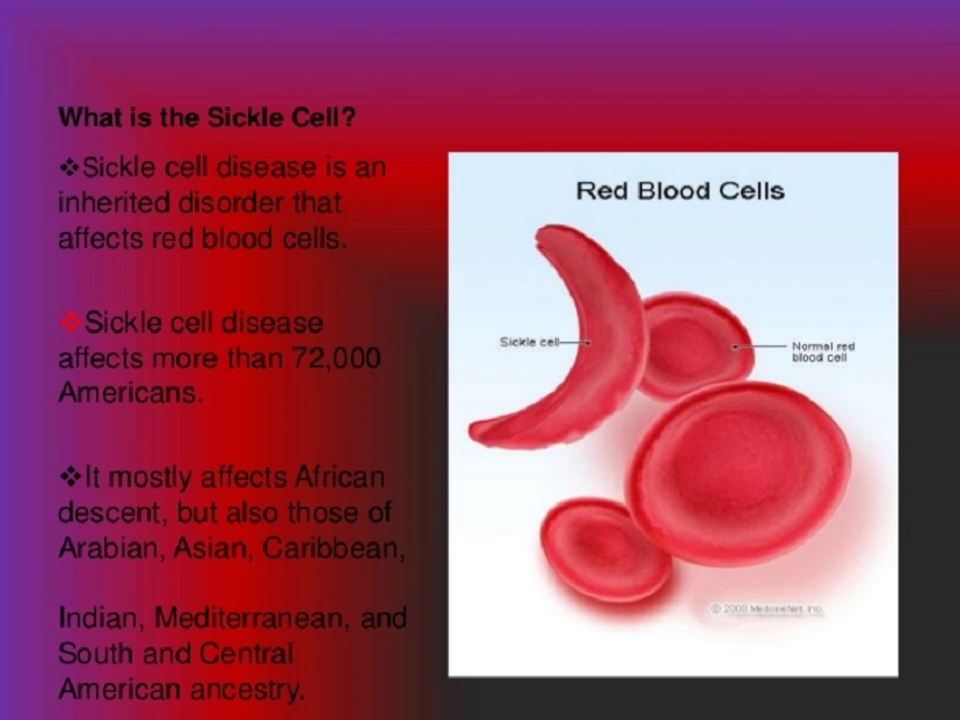
Sickle cell anemia is a genetic blood disorder that changes red blood cells from round to a rigid, sickle shape. Those misshapen cells clog small blood vessels, cause pain, and shorten red blood cell life. If you or someone you care for has sickle cell disease, the good news is there are clear steps to lower risks, treat crises, and improve day-to-day life.
Tell-tale signs and how it’s diagnosed
Common signs you’ll see: sudden, severe pain episodes (pain crises), chronic fatigue from anemia, frequent infections, swollen hands or feet in babies, and breathing trouble like acute chest syndrome. Some people also have delayed growth, vision changes, or stroke risk. Diagnosis is usually a simple blood test—newborn screening picks it up early in many countries. If there’s any doubt, doctors use hemoglobin electrophoresis to confirm the sickle hemoglobin type.
Treatment options
Treatment depends on age and symptoms. Hydroxyurea is the foundational drug for many patients — it reduces pain crises and hospital visits by raising fetal hemoglobin. For severe anemia or some complications, blood transfusions help quickly restore oxygen-carrying capacity. Repeated transfusions may require iron chelation to prevent iron overload. For acute pain, doctors use fluids, oral or IV pain meds, and oxygen when needed.
Curative options exist: bone marrow (stem cell) transplant can cure sickle cell disease for some patients if a matched donor is available. Newer gene therapies are emerging and show promise, but they’re not yet routine for everyone. Talk openly with a hematologist about whether a transplant or trial is an option for you.
Vaccines and antibiotics matter. People with sickle cell disease have higher infection risk, especially from encapsulated bacteria. Keep vaccines up to date and follow pediatric penicillin recommendations when applicable.
Hydration, temperature, and prevention help a lot. Drink water regularly, avoid extreme cold or high altitude when possible, and treat infections quickly. Those small habits cut the number of crises for many people.
Create a pain action plan with your care team. Know which over-the-counter meds are safe, when to use stronger prescription pain relief, and when to head to urgent care. For parents: teach older kids to recognize early signs of a crisis so they can act fast.
Watch for emergency signs: fever above 38.5°C (101.3°F), severe chest pain or shortness of breath, sudden weakness or slurred speech, or intense unrelieved pain. These need immediate medical attention.
Living with sickle cell disease is a daily challenge, but coordinated care makes a big difference. Find a hematologist experienced in sickle cell care, keep regular checkups, ask about hydroxyurea and transfusion strategies, and consider mental health support. With the right plan, many people lead active, productive lives despite the disease.
Sickle Cell Anemia and Dental Health: Protecting Your Smile
As a person living with sickle cell anemia, I've learned that dental health plays a crucial role in managing the disease. Our condition makes us more susceptible to dental problems, such as gum disease and tooth decay, due to reduced blood flow and slower healing. To protect our smile, it's essential to maintain a strict oral hygiene routine, including regular dental check-ups, brushing and flossing daily, and using a fluoride toothpaste. Additionally, staying hydrated, eating a balanced diet, and avoiding tobacco products are vital steps to keeping our teeth and gums healthy. By taking these precautions, we can minimize the risk of dental complications and maintain a beautiful smile.